Humoral rejection of the liver has recently been defined by the WORLD CONGRESSES OF GASTROENTEROLOGY consensus document(3) as "a relatively uncommon form of allograft injury and subsequent dysfunction, primarily mediated by antibody and complement, occurring immediately (hyperacute) or during the first week (acute) after transplantation. The antibodies are either preformed antibodies or represent anti-donor antibodies that developed after transplantation." This panel(3) considered acute humoral rejection, antibody mediated rejection and hyperacute rejection as acceptable synonyms.
Historically, it has been difficult to attribute liver allograft damage to preformed antibodies(4-9). This was largely because of a well-documented resistance of the liver to humoral rejection in comparison to kidney or heart allografts(2, 4-13). In addition, many "non-immunologic" complications, such as preservation injury and sepsis, can also produce a clinicopathologic syndrome quite similar to humoral rejection (14-16). Comparatively poor survival rates early after liver transplantation during the advent of clinical liver transplantation also made it difficult to correctly identify those who died or required retransplantation because of antibody mediated injury(16). With improvements in immunosuppression, patient selection, operative techniques and patient management, it is now clear however, that antibodies directed against the major ABO blood group antigens(14, 17-19) and MHC antigens(1, 20-26) can cause allograft failure, albeit rarely in a "hyperacute" fashion.
The first system to be described that predictably resulted in humoral hepatic allograft rejection was the major ABO blood group isoagglutinins(14, 17-19). Once this barrier was recognized, crossing the ABO blood groups in liver transplantation was generally avoided and humoral rejection because of a major blood group incompatibility became rare. However, in candidates with fulminant hepatic failure where the need is urgent or in children, where the donor pool is limited, preconditioning of an ABO incompatible recipient has been used. Various combinations of splenectomy, cytoreductive therapy, prophylactic anti-lymphocyte globulins(27), OKT3(28, 29) and pre- or post-transplant plasmapheresis(27) have yielded acceptable graft and patient survival results(30). Therefore, while ABO incompatibility poses a definite risk of humoral and acute cellular rejection, it is not considered to be an absolute contraindication to transplantation by some groups(29).
The potential of lymphocytotoxic antibodies, which are primarily
of the IgG class, to cause liver allograft damage and failure
has been more recently recognized by several centers(1, 23-26,
31-33), but not by others(31, 34, 35). Thus, there is no general
agreement therefore, on the importance of a prospective crossmatch
in liver transplant recipients(35). However, even though lymphocytotoxic
antibodies do not usually precipitate hyperacute rejection and
are clearly less destructive than the isoagglutinins in clinical
practice, data from experimental animals clearly show that antibody-mediated
rejection alone, or in combination with acute cellular rejection
significantly contribute to liver allograft damage (1). Thus,
knowing that the recipient harbors potentially destructive preformed
antibodies is likely to be helpful for patient management in the
early post-transplant period.
Pathophysiology
Antibodies directed at antigens expressed on the vascular endothelium are potentially the most destructive, since vascular injury interferes with the blood supply(1). Antibodies included in this group are those reactive with the major ABO blood group and class I MHC antigens, detectable in conventional blood typing and lymphocytotoxic crossmatch tests, respectfully. In xenotransplantation, polysaccharide antigens on the surface of endothelial cells are a major barrier to successful engraftment(36, 37).
Studies in experimental animals have provided important information about the mechanisms of enhanced allograft damage in presensitized recipients. Qian et al(38) have shown in experimental animals that MHC class I rather than class II molecules play an important role in allosensitization. The presensitization is likely related to an augmentation of the TH1-type cytokines and immune responsiveness(39). These observations are consistent with the clinical findings that alloimmune antibodies of the IgG class are the most destructive(2, 40).
The critical event in the effector phase of humoral rejection appears to be antibody binding to the endothelium and subsequent complement fixation and activation. This results in direct endothelial damage, the formation of platelet-fibrin thrombi, initiation of the clotting cascade, subsequent microvascular thrombosis and arterial vasospasm, all of which act in concert to ruin the microvasculature, impair blood flow and eventually cause hemorrhagic necrosis.
The well-known resistance of the liver to humoral rejection has provided important insights about the pathophysiology of humoral rejection. Secretion of soluble MHC Class I antigens by the liver, Kupffer cell phagocytosis of cytotoxic antibodies, complement, immune complexes and activated platelet aggregates; the dual hepatic blood supply through the hepatic artery and portal veins and the unique hepatic sinusoidal microvasculature, which is devoid of a conventional basement membrane(1, 10, 11, 13, 41-45) have all been cited as explanations for the liver's ability to withstand the impact of pre-formed anti-donor antibodies much better than other organs. Recently, observations in liver xenotransplantation have shown that compatibility of complement components is yet another explanation for the hepatic resistance to humoral rejection(36, 46). It is known that complement mediated lysis of a target is less effective if the target cell and complement are derived from the same source (donor versus recipient species), than if they are from different sources. Thus, by providing syngeneic complement, a liver may protect it's own endothelial cells from the complement mediated lysis triggered by the preformed antibodies(36, 46). Whether this form of protection is operable in allografts has yet to be determined with certainty.
Even though all of these mechanisms can protect a liver allograft from antibody mediated damage, Knechtle et al(47), Gubernatis et al(48) and others(2) have clearly shown in animals that they could be overridden by intense presensitization. Nakamura et al(44) have recently outlined the events of humoral rejection in a clinically relevant small animal model. Some of the earliest morphological signs of humoral rejection are the presence of platelet-fibrin thrombi in the sinusoids , portal and central veins. These appear within one to three minutes after transplantation and are even better illustrated in plastic-embedded sections and through ultrastructural analysis. Within one to three hours, endothelial hypertrophy and vacuolization, spotty acidophilic necrosis of hepatocytes, and neutrophilic portal venulitis appear. In 6-24 hours, small areas of hepatocyte necrosis are evident and can become confluent. If the preformed antibodies are high titer, IgG and show endothelial specificity(2), then rapid graft failure from widespread hemorrhagic necrosis can occur. However, if the protective mechanisms, described above, can override the damage, gradual resolution of the platelet-fibrin thrombi occurs during the next three to twenty four hours after transplantation.
If the allograft does not precipitously fail, acute (cellular) rejection frequently develops earlier than it normally would, in non-sensitized recipients.
Clinical Presentation
The International Panel referred to earlier describes humoral
rejection as severe allograft dysfunction without an obvious cause
occurring in a presensitized patient immediately after or during
the first week after transplantation(3). The allograft may become
swollen, cyanotic, and mottled; bile production may slow or stop.
Establishing the diagnosis requires that conditions such as
"preservation"
injury, hepatic artery or portal vein thrombosis, or venous outflow
obstruction can be excluded with reasonable certainty. Depending
on the severity of injury, the above findings may be accompanied
by a consumptive coagulopathy and operative site bleeding, with
a need for blood components(3).
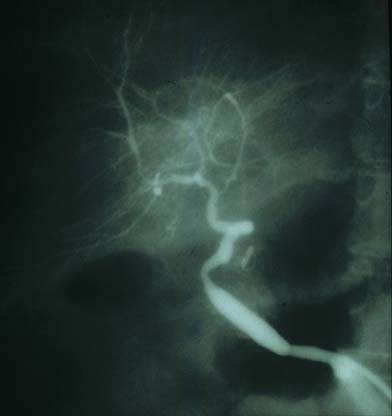
ABO Incompatible: The first signs of serious liver injury often develop in the operating room after vascular re-anastomosis and before abdominal closure(14, 18, 19, 49, 50). The liver usually reperfuses uniformly and produces bile, but within minutes or hours becomes hard and swollen before bile flow slows or stops altogether. An inordinate need for platelets and difficulty in achieving hemostasis signal the initiation of an intrahepatic consumptive coagulopathy (40). However, the intra-operative events are rarely serious enough to abort the procedure or undertake immediate retransplantation. An unexplained rise in liver injury tests during the first several post transplant days, refractory thrombocytopenia, hypocomplementemia and symptoms signal the possibility that humoral rejection is occurring(14, 19, 40, 49, 50). At this point, hepatic angiography is often obtained to investigate the cause of the unexplained allograft dysfunction. In the typical case, it shows segmental narrowing, or a "sausage-link" appearance(14, 19, 21, 49, 50) and/or diffuse luminal narrowing with poor peripheral filling. These are signs indicative of immunologically-mediated arterial vasospasm.
Unfortunately, in conventionally treated recipients of ABO incompatible
organs the marked rise in transaminases is followed in 60% - 70%
of cases by synthetic function failure, subsequent wound site
bleeding and other systemic signs of hepatic failure, that necessitate
retransplantation(14, 19, 21, 49, 50). Those that survive the
early insult are more prone to the development of biliary tract
strictures late after transplantation(14, 51).
ABO Compatible: Lymphocytotoxic antibodies in general, cause less serious injury than the isoagglutinins(1, 20-26, 40). In addition, the ability of various lymphocytotoxic antibodies to effect graft damage greatly varies, which appears to be related to the antibody titer, specificity and class(2, 40, 44). The IgG class reportedly cause the most damage(1, 20-26, 40). In addition, one cannot exclude the possibility that the use of blood products containing anti-donor antibodies and active complement can contribute to the injury.
In general, the higher the titer of IgG anti-MHC antibodies detected on the routine crossmatch before transplantation(23, 25, 26) the more likely the patient will encounter significant difficulties during and after the operation(2, 40, 44). However, the relative risk to the recipient and the potential number of patients involved should be kept in perspective. Crossmatch positivity for IgG lymphocytotoxic antibodies is typically encountered in 8 -12 % of all liver allograft recipients, and of those, only 30% have more dangerously high titers(40). Thus, the patient population at greatest risk is relatively small (23, 26, 40) and in a liver transplant program where there are fewer than 100 cases per year, humoral rejection may be overlooked as a cause of dysfunction or failure.
The most frequent clinical presentation is a persistent rise in
serum bilirubin that occurs during the first week after transplantation,
accompanied by refractory thrombocytopenia(52), low complement
activity, and a biopsy showing changes of "preservation injury"(40).
This is usually followed by the onset of
acute (cellular) rejection and the need for increased
immunosuppression(1, 2, 44).
Ischemic biliary necrosis later manifest as
biliary sludge,
obstructive cholangiopathy and small bile duct loss are other serious late
manifestations(1, 2, 44, 51, 53). In rare cases, precipitous hemorrhagic
necrosis similar to that seen with isoagglutinins can occur(32).
Gross and Histopathology
The diagnosis of humoral liver allograft rejection is difficult to establish with certainty, and implies that other causes of early liver allograft failure such as preservation injury, vascular compromise, sepsis and trauma have been reasonably excluded. This usually requires a "backtable" liver biopsy that shows no significant steatosis, portal inflammation, hepatocyte necrosis or immunoglobulin deposits.
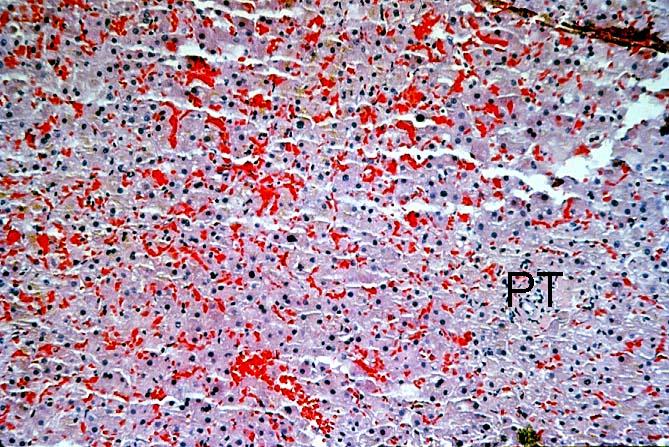
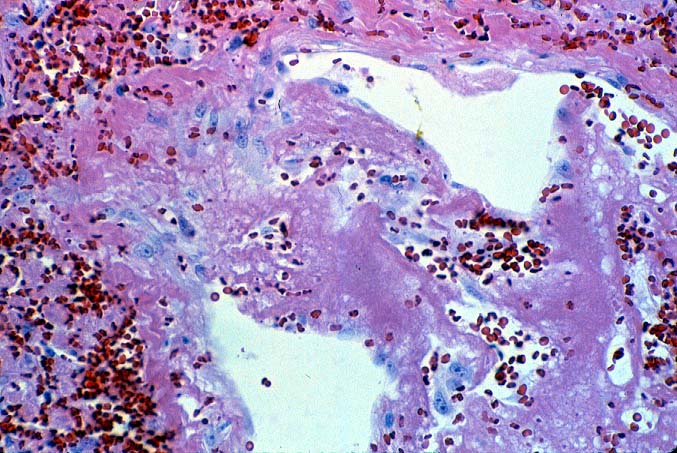
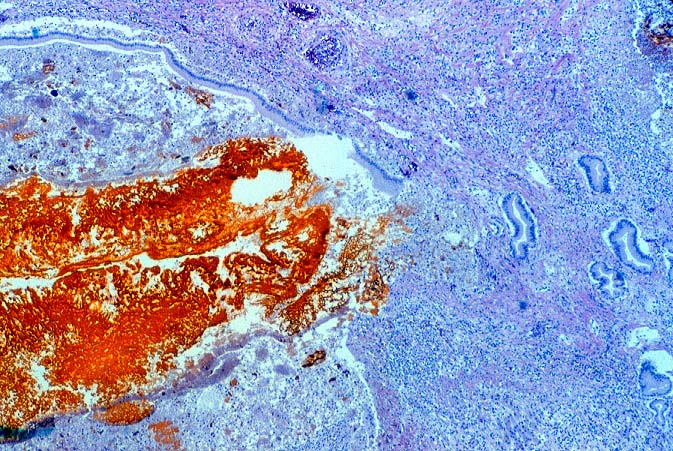
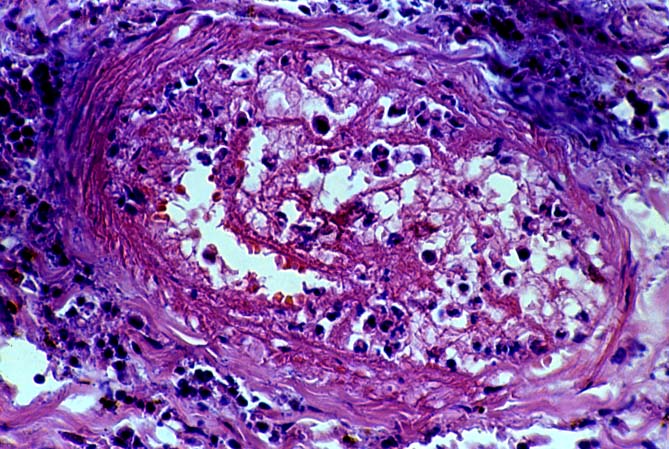
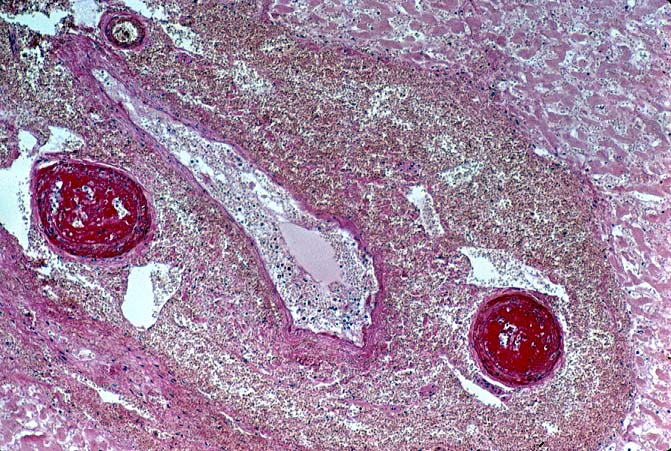
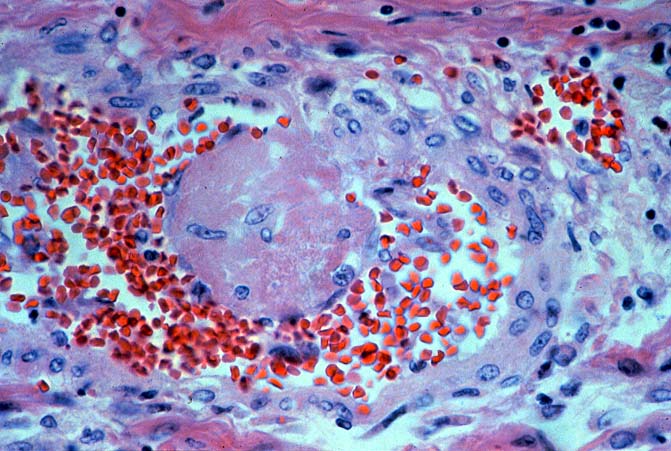
ABO Incompatible Organs: The histopathologic findings depend on the timing of the biopsy and the severity of the injury. Samples taken immediately after reperfusion from patients with marked injury may show an impressive sludging of red blood cells and a clustering of neutrophils in the sinusoids. Focal platelet-fibrin thrombi in portal and central veins can also be seen. Hemorrhage into the space of Disse', small areas of ischemic hepatocellular necrosis usually follow within the next hours to days(14, 19, 49, 50).
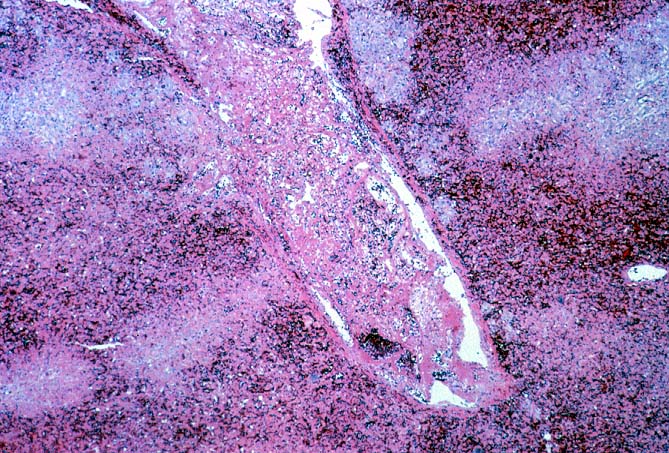
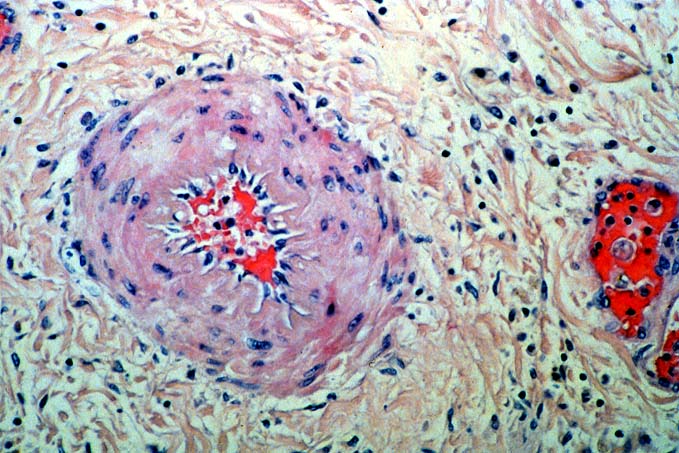
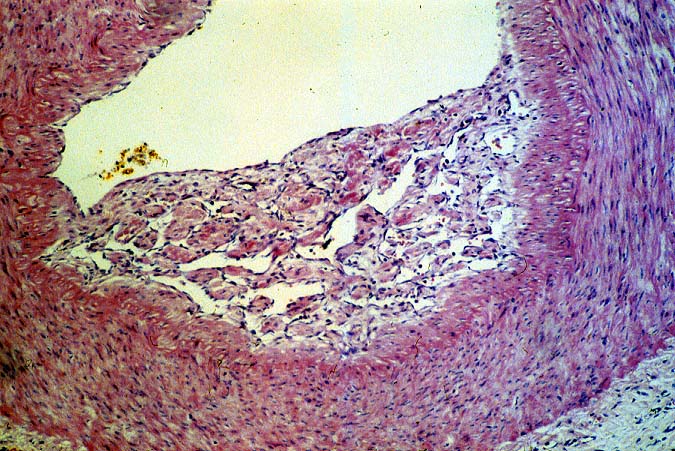
Biopsies taken within the first several days in those with severe injury will show confluent coagulative hepatocyte necrosis without any particular lobular distribution. Nearby portal veins contain circumferential fibrin deposition and markedly hypertrophic endothelial cells. Arteries are usually less severely affected, although neutrophilic and/or necrotizing arteritis can be seen on occasion. A mild neutrophilic portal exudate usually appears at 2 to 3 days, as does focal cholangiolar proliferation. The latter finding is interpreted as a hepatic response to injury in the periportal region. Thereafter, progressive hemorrhagic infarction of the organ occurs in patients destined for allograft failure(14, 19, 49, 50).
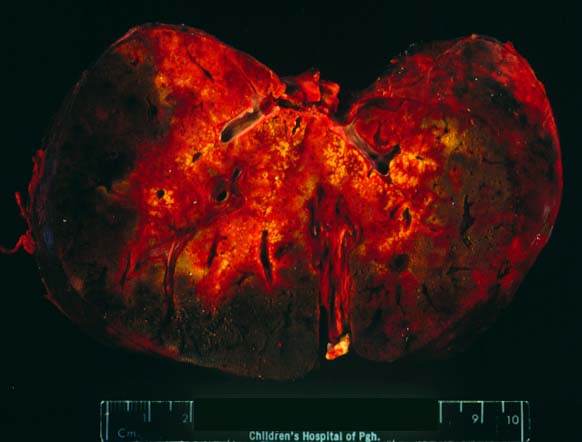
Failed ABO incompatible allografts examined at the time of retransplantation often reveal enlarged, cyanotic organs, mottled with areas of necrosis(14, 49), with or without rupture of the capsule. Hepatic artery and portal vein thrombosis is variably present. Microscopically, large geographic areas of hemorrhagic necrosis are the most frequent findings. On closer examination, focal fibrinoid necrosis of arteries may be seen but is present in only a minority of cases. More common vascular findings include arterial and venous endothelial cell hypertrophy, neutrophil sludging, focal fibrin deposition around a partial circumference of the vessel, with a mass of fibrin extending into the lumen (14, 49). The presence of arterial medial thickening and myocyte vacuolization are common and probably represent morphologic manifestations of arterial vasospasm. In addition, a rapidly developing fibro-intimal hyperplasia can occur. Occasionally, if the allograft survives the first week after transplantation the injury will subside. However, there is an increased risk of long term biliary tract strictures, presumably related to residual arterial pathology, such as re-canalized thrombi.
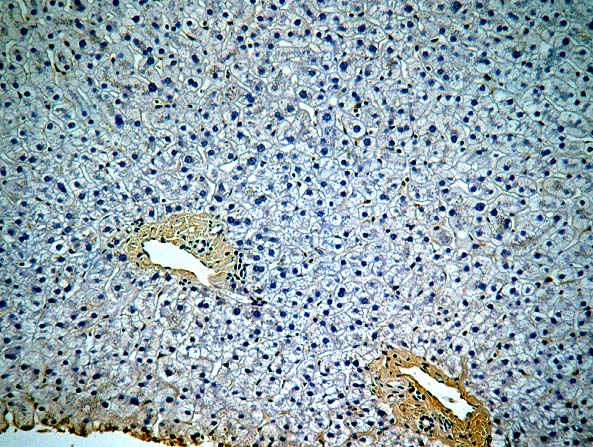
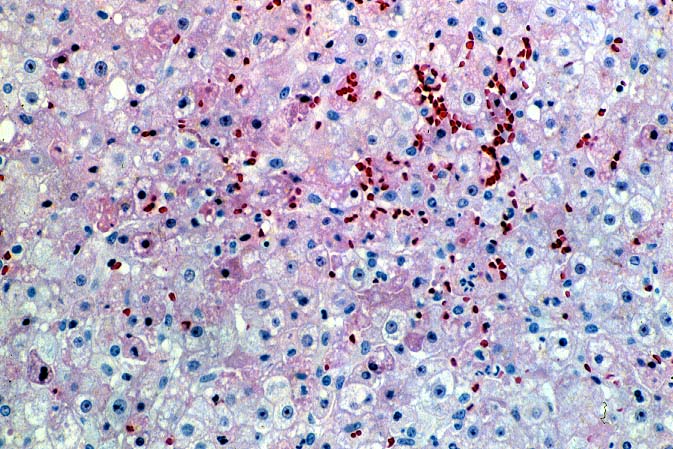
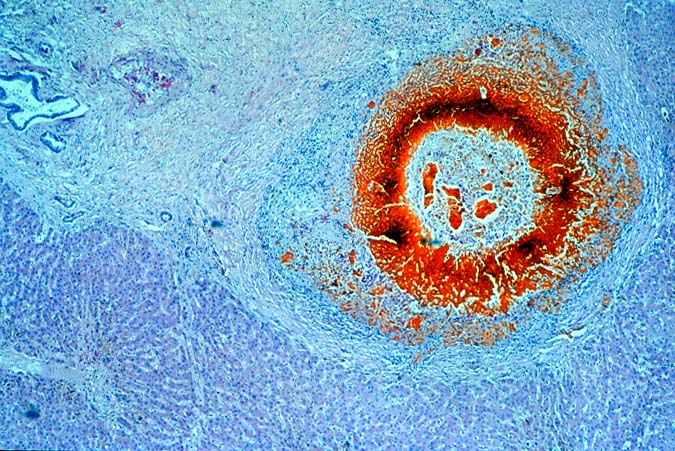
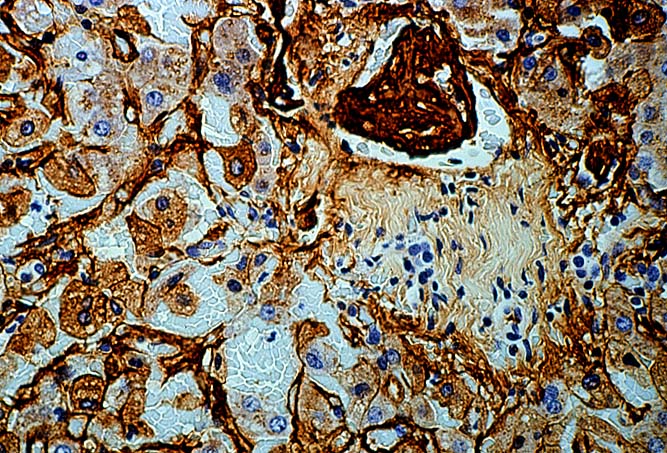
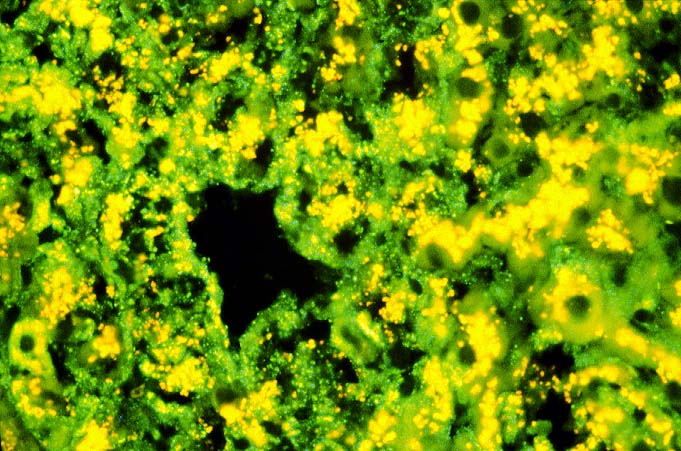
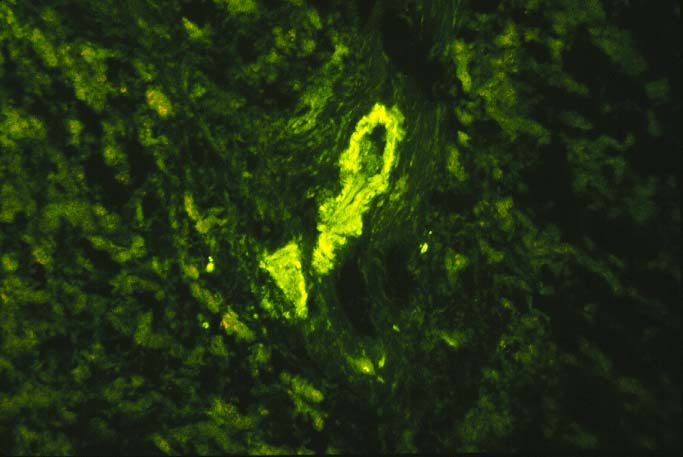
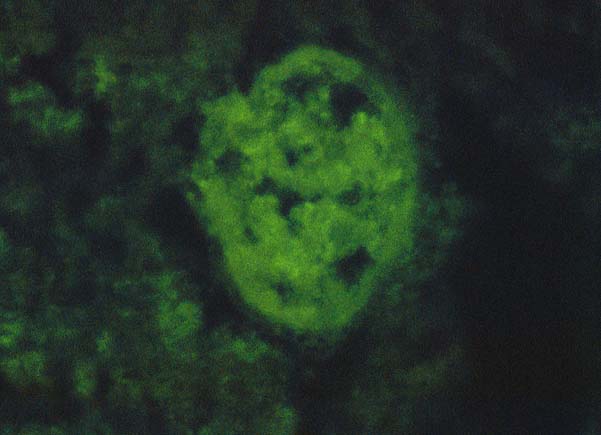
Immunofluorescent
and immunoperoxidase stains in
ABO incompatible organs will often reveal IgM, focal IgG, C3, C4 and Clq
in an occasional artery,
in the hilar microvasculature and in the
sinusoids.
However, arterial deposits of IgG, IgM and Clq may be seen in
a similar distribution in allografts with non-immunologically
mediated injury. Background fluorescence
in the portal connective tissue and sinusoids can present a problem
during interpretation of the findings. Elution studies can be performed to
confirm the identity of the deposited antibodies(14, 49). The final diagnosis
should be based on a complete clinicopathologic analysis during
which other non-immunologic causes of graft failure are reasonably
excluded.
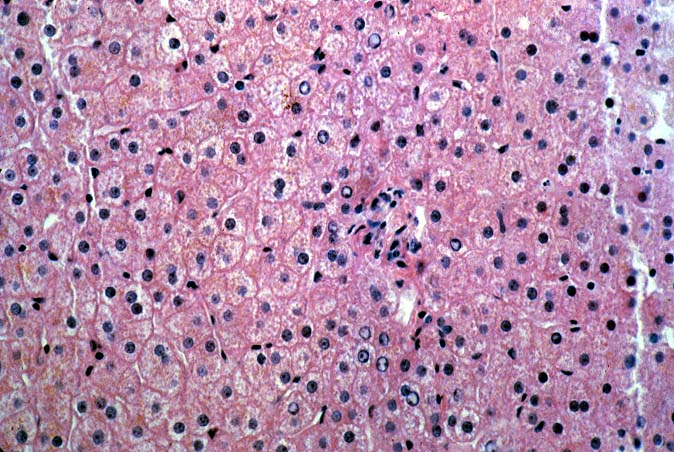
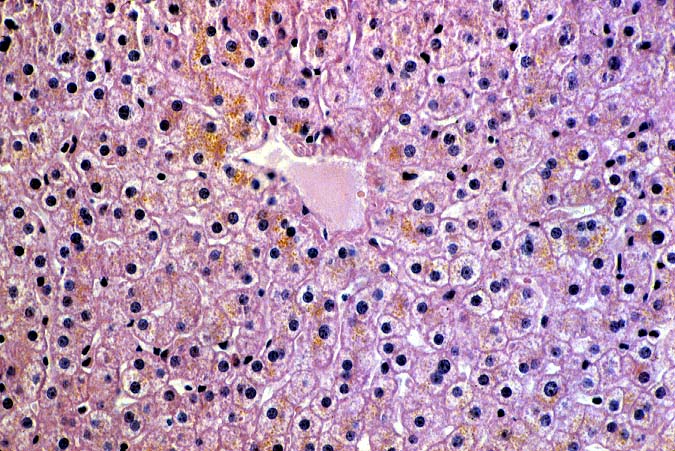
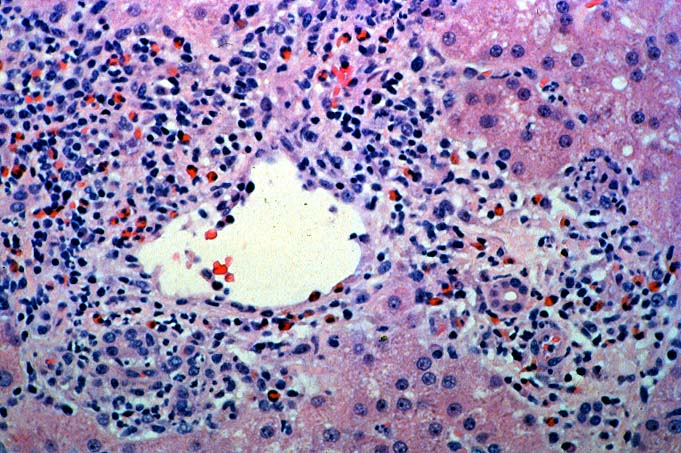
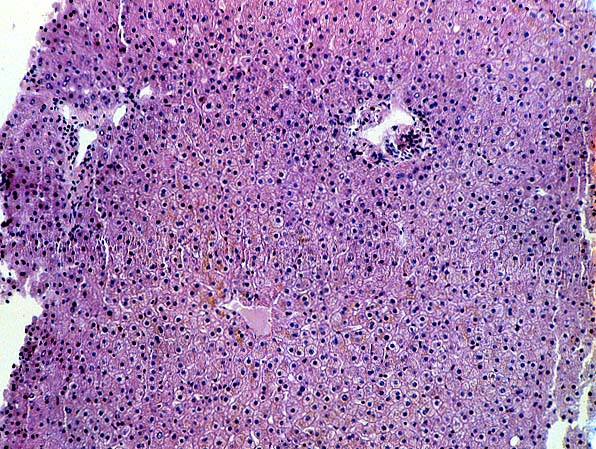
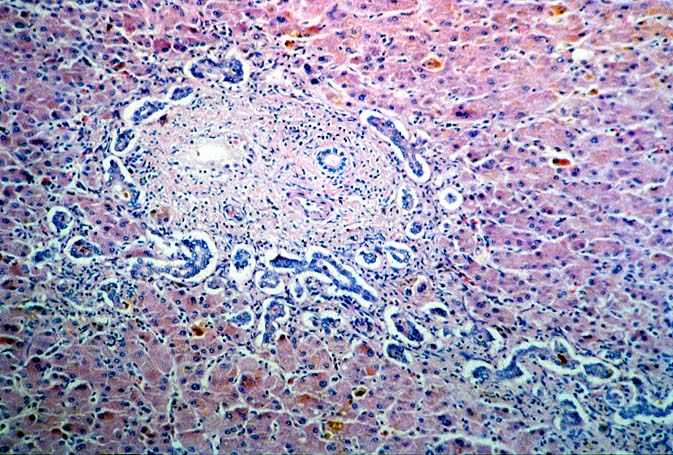
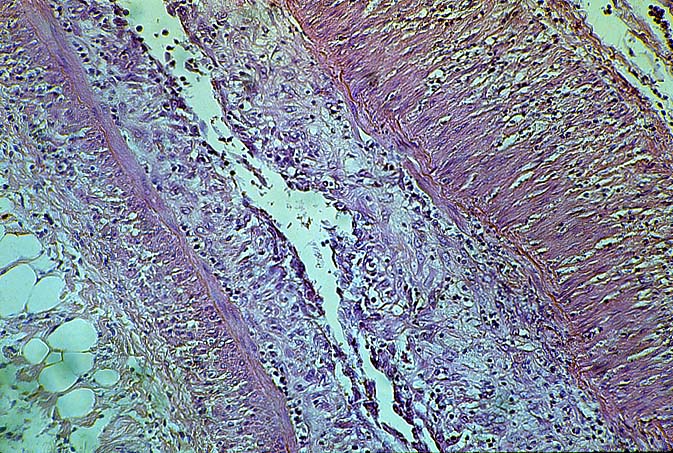
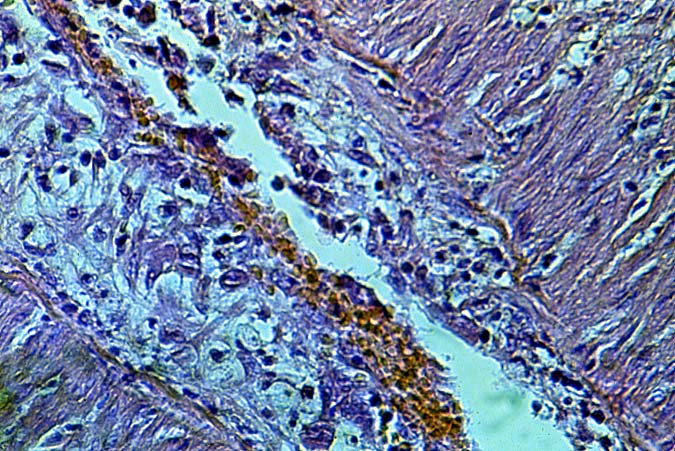
ABO Compatible Organs: In general, the hepatic damage is more variable and generally less severe in patients with a positive crossmatch than in those with incompatible isoagglutinins. In addition, the injury pattern is more often difficult to separate from other insults that mimic humoral rejection. In the typical clinical case, reperfusion biopsies from patients with a positive lymphocytotoxic crossmatch more often contain platelet aggregates in the portal and or central veins than crossmatch negative controls(1, 44). Spotty acidophilic necrosis of hepatocytes, centrilobular hepatocellular swelling, accompanied by cholangiolar proliferation and hepatocanalicular cholestasis often appear during the first week after transplantation. Neutrophilic or necrotizing arteritis is rare(1, 44) in needle biopsy specimens, but lymphocytic subendothelial infiltration is not uncommon in large hepatic artery branches. Overall, the histopathologic changes closely resemble those of "preservation" injury, except for subtle arterial changes, which also may not be present in needle biopsy samples. These are best observed in arteries from the perihilar region of allograft hepatectomy specimens and include endothelial hypertrophy, lymphocytic arteritis, medial thickening, partially organized thrombi, necrosis of individual myocytes, and medial myocyte vacuolization(1, 44). Other hilar changes in humoral rejection include congestion of the peribiliary vascular plexus and biliary necrosis of large septal bile ducts(1, 44).
If allograft failure does not occur in a positive crossmatch patient, acute rejection , manifest as cellular infiltration of the liver, usually becomes evident within 5 - 7 days of transplantation(1, 44). This makes the cause of injury and dysfunction more obvious. If the allograft survives the early post-operative injury, long term sequela of an early humoral insult either from isoagglutinins or lymphocytotoxic antibodies can include: biliary sludge and stricturing with obstructive cholangiopathy, and obliterative arteriopathy and loss of small bile ducts, or chronic rejection(14, 19, 27, 51).
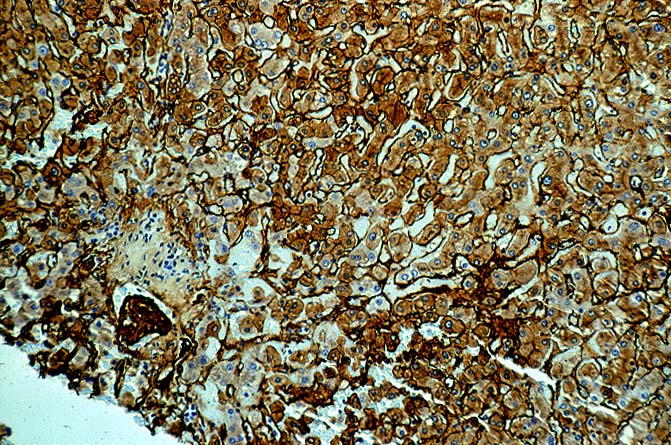
In our experience, the immunofluorescence findings in ABO compatible allografts can be supportive of humoral rejection, but alone, they rarely are diagnostic unless the immune deposits are intense. Deposits of IgG, C3 and C4 in the arteries and in the portal and hilar microvasculature, without heavy a-2-macroglobulin or other macromolecules suggest specific deposition. Therefore, such deposits are more indicative of humoral rejection than are "non-specific" localization of IgM, Clq and other macromolecules, which frequently become lodged in necrotic arterial walls, regardless of the cause of damage.
The International Panel suggests that the minimal diagnostic
criteria are: rapid onset liver dysfunction with histologic
features of ischemic necrosis and predominantly neutrophilic infiltrates,
in the absence of other clearly defined causes of ischemia or
infarction. The diagnosis is strengthened if neutrophilic or necrotizing
arteritis is present, if immunoglobulin deposits can be demonstrated
in the liver, and if preformed anti-donor antibodies are found.
Technical and preservation-related causes of ischemia infarction
should be reasonably excluded(3).
Differential Diagnosis
Severe humoral rejection can easily be confused with other insults that cause hemorrhagic hepatic necrosis, such as severe hypotension, sepsis or vascular thrombosis(1, 14, 15, 49, 54). Reconstructing the clinical course of events is made easier if one is aware of the ABO compatibility and crossmatch status. Under ABO-compatible circumstances, humoral rejection is most often confused with "preservation" injury and the pre-sensitization state and clinical profile can provide information useful for the interpretation of the histopathologic findings. For example, a recipient that harbors high titer (> 1:32-500) preformed IgG lymphocytotoxic anti-donor antibodies, should be assumed to be at greater risk for humoral rejection, compared to a recipient with low titer (< 1:32) anti-donor antibodies. The diagnosis of humoral rejection is strengthened when there is no obvious technical or other cause of allograft dysfunction(23, 26, 40), and if there is persistence of the preformed antibodies after transplantation. In addition, an antibody insult is usually accompanied by a drop in the platelet count and persistent thrombocytopenia below 50,000, and hypocomplementemia, as compared to normal pretransplant values(40).
The clinical findings should be interpreted in conjunction with histopathologic findings on liver biopsy. Histopathological clues that humoral rejection may be occurring include arterial endothelial cell hypertrophy, arterial medial thickening and myocyte vacuolization associated with features suggestive of parenchymal ischemia, such as centrilobular hepatocellular swelling or frank necrosis(1). The more definite histopathologic features of humoral rejection, such as lymphocytic arteritis and fibrinoid necrosis, are rarely seen in a peripheral needle biopsy. With severe preservation injury the cholangiolar proliferation, acute cholangiolitis, centrilobular swelling and hepatocanalicular cholestasis generally improve over time. These same changes generally worsen with time in patients with humoral rejection, unless additional immunosuppression is given.
Recently, a syndrome of fever and sudden deterioration of graft
function which mimics humoral rejection has been seen in association
with high levels of interferon- and tumor necrosis factor-, but
without preformed antibodies(55). Other groups have observed progressive
deterioration of allograft function associated with severe microvascular
steatosis on light microscopy that was could not be attributed
to a specific cause(56).
REFERENCES
|
|
|
|