

The highly infectious nature of HBV and the presence of extrahepatic reservoirs of virus probably account for the difficulty eradicating the virus before transplantation and its high recurrence rate after transplantation. Nevertheless, various treatment modalities have been used in an attempt to break this disheartening cycle of re-infection and recurrent disease, which all too often, leads to allograft failure and/or death. These include hepatitis B immune globulin, various anti-viral drugs(17), active vaccination with HBsAg, alpha-interferon (8, 18, 19), human anti-HBs monoclonal antibody and the use of baboon liver donors for transplantation, which are resistant to type B viral hepatitis infection(20, 21). Except for xenotransplantation(21), which is not yet a viable treatment alternative for most patients, none of the above therapies can prevent re-infection. However, continuous high dose anti-HBs therapy has been able to delay the onset and ameliorate the injury associated with recurrence of the B virus after transplantation(11, 14, 18, 22-26).
Since significant recurrent disease is a problem for most HBV+ recipients,
several programs questioned even whether liver transplantation
should be carried out in patients who are infected with the B
virus(8, 9, 27, 28). HBV+ recipients also have a high incidence
of complicating hepatocellular carcinomas that have the potential
to contribute to diminished patient and graft survival after transplantation(8, 29). Detailed studies however,
show that recurrence of the viral infection, and not the cancer, is the
major cause of morbidity and mortality after transplantation in HBV+ patients with co-existent hepatocellular carcinomas(8, 30). Moreover, encouraging reports
of the ameliorating effect of specific anti-viral therapy on disease
progression after transplantation have tempered the pessimism
such that many centers continue to offer liver transplantation
to patients infected with the B virus.
Pathophysiology
It is not our intention to engage in a detailed discussion of the pathophysiology of hepatitis B virus-induced liver disease. However, an overview of some of the pathophysiologic mechanisms of HBV-induced liver disease may help to explain some of the histopathological manifestations of HBV in an allograft liver. Under normal circumstances, the hepatitis B virus is not thought to be cytopathic: liver damage is thought to be at least partially attributable to the expression of hepatitis B core antigen on the surface of hepatocytes, which then become targeted for destruction by MHC-restricted cytotoxic T lymphocytes (CTL)(31, 32). Thus, expression of viral antigens on the surface of hepatocytes precipitates lymphohistiocytic lobular lobular inflammation and spotty acidophilic necrosis of hepatocytes, typical of acute hepatitis. IN the general population, this is followed by "clearance" or control of the virus in most instances, or evolution toward chronic disease in a minority of recipients. In a liver allograft recipient, potent immunosuppression needed to prevent rejection interferes with normal mechanisms of viral clearing and contributes to the progression to chronic disease in most recipients. In fact, lower maintenance doses of corticosteroids may modify the course of post-transplant HBV infection by leading to less viral replication, milder HBV-related liver disease and better overall patient survival(33). In addition, since there is no attempt to prospectively match the donor and recipient for MHC antigens, normal pathways of CTL lysis of virally infected hepatocytes may also be disrupted. Instead, viral could be processed by recipient accessory cells and presented to T lymphocytes in a MHC-class II restricted fashion, resembling a delayed type hypersensitivity response(3, 34).
Although MHC mismatching has the potential to significantly disrupt the manifestations of HBV-related disease in a liver allograft, there are too few cases at this time to draw any definite conclusions about how this actually occurs. Nevertheless, massive viral replication in class II MHC mismatched deteriorating liver allografts with little or no hepatic inflammation, prompted several groups, including our own, to suggest that, under special circumstances, the virus may be directly cytopathic(3, 5-7, 35, 36).
Clinical Presentation
HBV hepatitis usually first appears between six to eight weeks after
transplantation. Because liver injury tests are routinely monitored in this population, an elevation of the ALT and AST usually first prompts suspicion of recurrent disease. However, more dramatic initial presentations include nausea, vomiting, jaundice and hepatic failure, similar to that seen in the general population or in other immunosuppressed patients(1-12). The diagnosis is suspected on clinical grounds and confirmed after examination of a core needle biopsy.
Histopathologic Findings
Although local treatment policies can influence the timing and severity of the histopathologic changes, in general, HBV hepatitis in liver allografts is similar to that seen in non-allograft livers(1-10, 12, 35, 36). There is a typical progression from an acute hepatitis with predominantly lobular changes to chronic hepatitis, when a combination of portal tract and lobular changes are observed. Cirrhosis can develop with striking rapidity(1-10, 12, 35, 36). However, an occasional patient will show histopathologic resolution of disease activity after a bout of acute hepatitis, and rare patients will actually "clear" the virus after transplantation. There also are several somewhat unique histopathological presentations of HBV in the liver allograft recipient, that are likely related to the potent immunosuppression and MHC non-identity between the liver and recipient(see below).
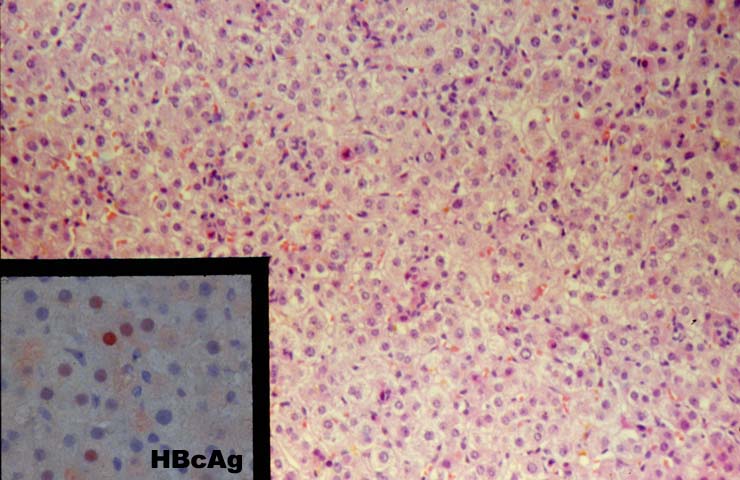
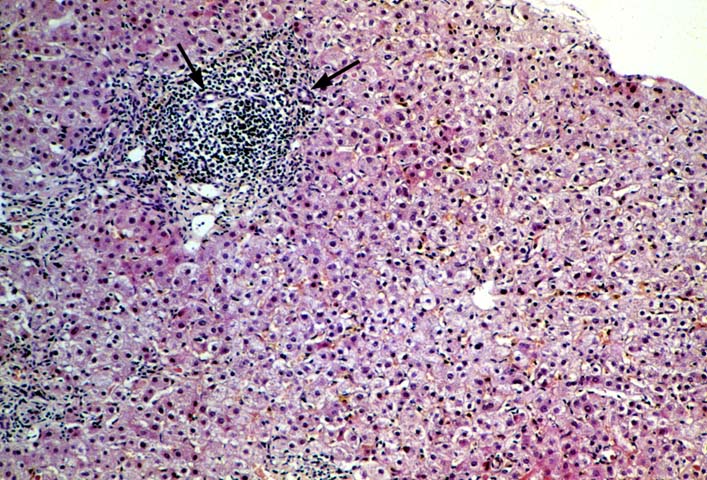
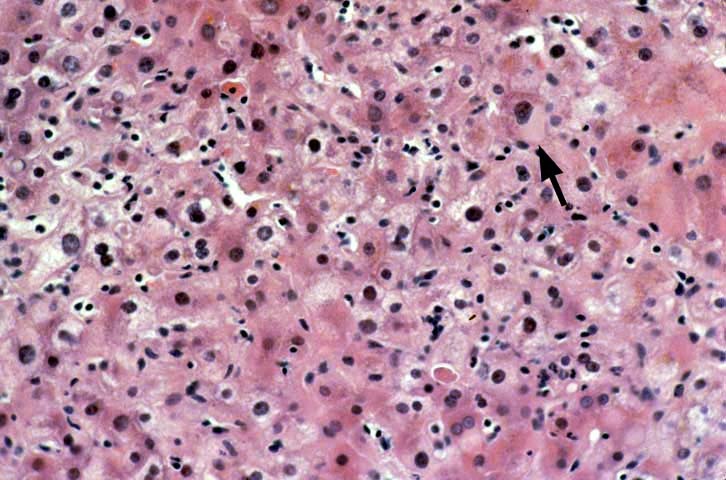
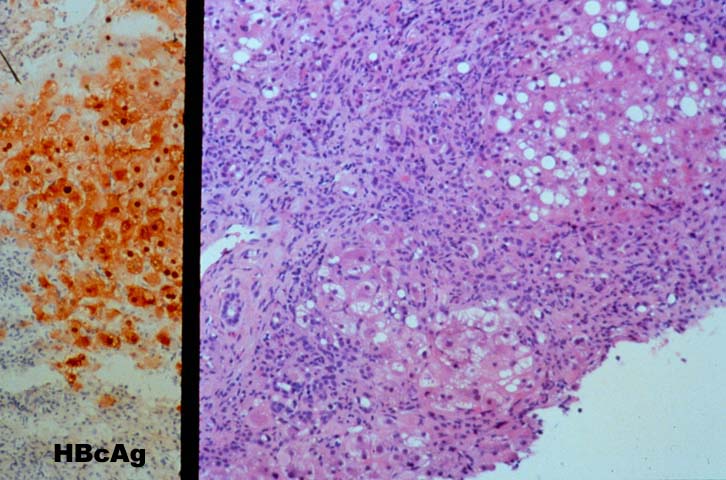
Re-infection with the type B hepatitis virus is usually first detected on immunohistochemical stains for HBV antigens. Within several weeks after transplantation, hepatitis B core antigen can appear in the cytoplasm of an occasional hepatocyte. The infection, and thus core and surface antigens, spread throughout the liver lobules to involve a larger number of cells(1-10, 12, 35, 36). This is followed by acute hepatitis in which the lobular changes predominate. These include lobular disarray, Kupffer's cell hypertrophy, inflammation and necro-inflammatory activity; and variable portal inflammation. At this stage of the disease, a small percentage of patients will quickly progress to bridging or even submassive necrosis, particularly if immunosuppression is withdrawn(1). However, most patients show some resolution of the acute lobular necro-inflammatory activity, but evolution toward chronic hepatitis. The later phase is marked by predominantly mononuclear portal inflammation and interface activity. The lobular changes become less pronounced.
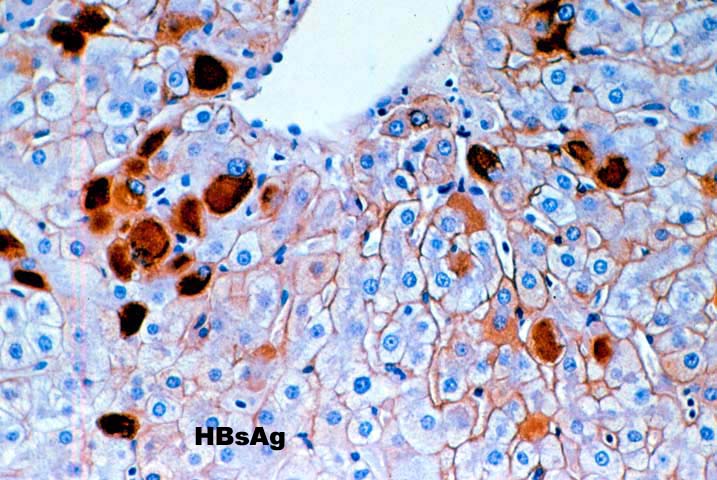
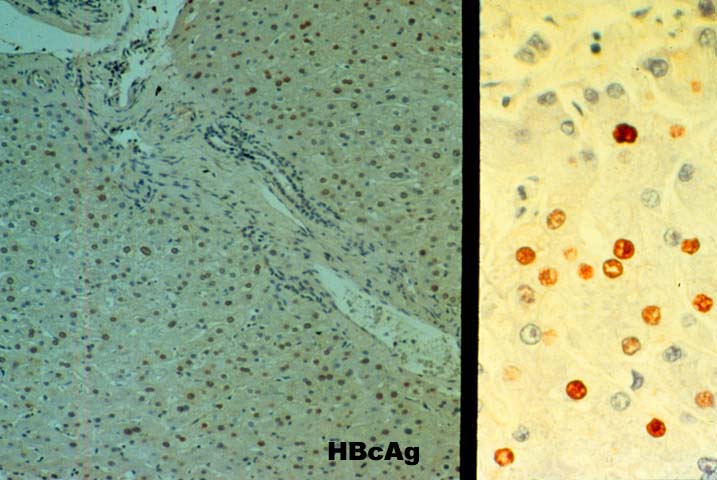
The typical case of chronic hepatitis in a liver allograft is similar to that seen in the general population and the same histological findings are used to ascertain the diagnosis. This includes lymphoplasmacytic portal inflammation that spares the bile ducts and portal veins, but shows interface activity or peri-portal hepatitis of varying severity, characterized by extension of lymphocytes and macrophages into the edge of the lobule combined with cholangiolar proliferation. Lobular findings seen during the chronic phase of the disease include a large number of cells with a ground glass cytoplasm containing hepatitis B surface antigen and, on occasion, numerous hepatocytes with sanded nuclei filled with hepatitis B core antigen. This is accompanied by varying degrees of lobular disarray, regeneration, Kupffer's cell hypertrophy and lobular necro-inflammatory activity.
Unique Histopathological Presentations
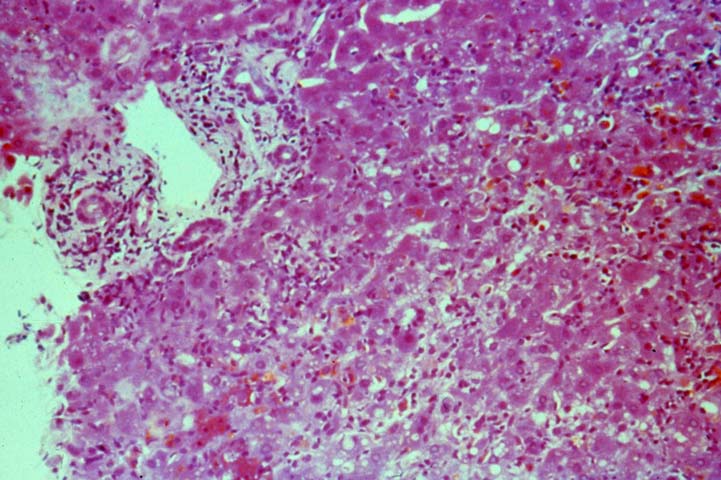
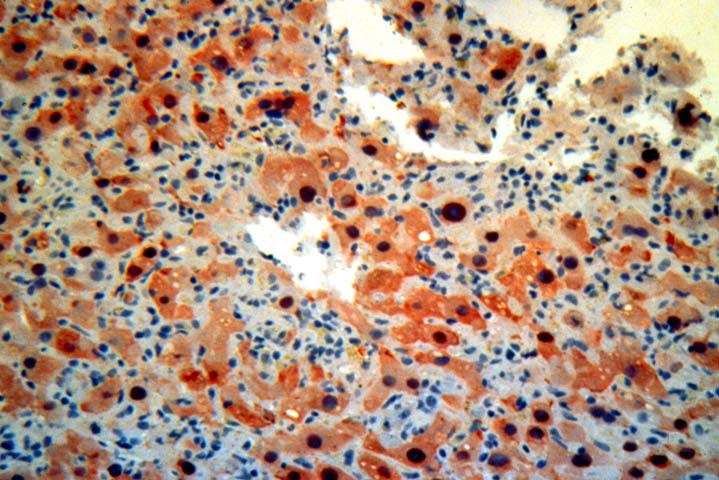
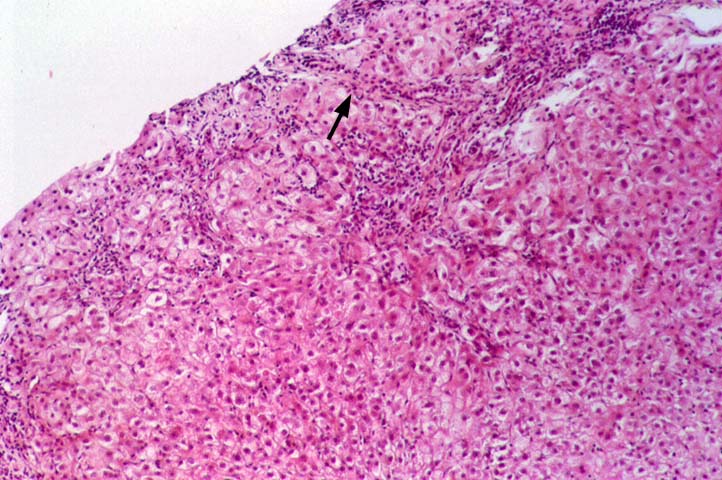
Several histopathological patterns of liver injury associated with HBV infection of a liver allograft are not commonly encountered in the general population. This is probably (but not definitely) related to effects of immunosuppression and MHC non-identity between the liver and recipient(3, 5-7, 10, 35, 36). The first unique histopathological pattern(3), termed "fibrosing cholestatic hepatitis (6)" is characterized by marked hepatocyte swelling, lobular disarray and cholestasis, combined with minimal or no portal or lobular inflammation, and cholangiolar proliferation of varying severity. Such cases are usually marked by massive hepatocellular expression of core and/or surface antigen, that apparently lead to the hepatocyte degenerative changes including cell swelling, steatosis and necrosis. Follow-up biopsies in such patients will often show progressive portal expansion because of cholangiolar proliferation, peri-portal sinusoidal fibrosis and lobular collapse, often without a significant inflammatory component(3, 5-7, 10, 35, 36). High levels of viral replication and antigen expression in these cases have led several groups to suggest that HBV may be directly cytopathic(3, 5-7, 10, 35, 36). Other less popular names that have been used to describe these constellation of findings, or ones very similar, include "fibrosing cytolytic hepatitis"(36) and "steatoviral" and "steatofibroviral" hepatitis (35) to emphasize the presence of hepatocellular cytolysis, steatosis and fibrosis, respectively.
Finally, a massive overproduction of the surface antigen can be seen in some patients. In these cases, the vast majority of hepatocytes contain a ground glass cytoplasm, similar to HBsAg transgenic mice(37). This results in marked distortion of the hepatocytes, lobular disarray and mild necro-inflammatory lobular activity.
HCV and Delta Co-infection
In patients who undergo liver replacement for simultaneous HCV
and HBV disease, the recurrent hepatitis after transplantation more frequently resembled recurrent HBV than recurrent HCV. (38). In addition, one group suggests that the presence of HCV may actually improve the clinical outcome of recurrent HBV as compared HBV infection alone (39).
As in the general population, co-infection with the delta agent can complicate
HBV disease in the liver allograft recipient. The consequences of delta agent co-infection are not entirely clear because of somewhat conflicting follow-up results. There have been reports of both more and less severe disease after transplantation(10, 12, 40-43) compared to recipients with HBV alone. This might be related to the cytopathic effect of HDV in liver allograft recipients in relation to HBV replication. David et al(44) have noted that in patients with non-replicative HBV infection, HDV hepatitis produced histopathological lesions similar to those described in fibrosing cholestatic hepatitis. However, prominent HBcAg expression was not seen in the hepatocytes. In contrast, when HBV is actively replicating, co-infection with HDV resulted in necro-inflammatory activity similar to that seen in B & D viral hepatitis in patients from the general population(44).
Differential Diagnosis
During the acute phase, viral hepatitis type B should be distinguished from acute hepatitis caused by other "hepatitic" viruses such as type C and from "opportunistic viruses in the Herpes group. All can show a similar histopathological pattern of injury which includes lobular disarray, Kupffer's cell hypertrophy, lymphohistiocytic lobular inflammation, spotty acidophilic necrosis of hepatocytes and variable portal inflammation. The presence of inclusion bodies or ground glass cells can be used to distinguish HBV from hepatitis caused by viruses in the Herpes group. However, ground glass cells are uncommon during the initial presentation. Therefore, the distinction is usually made with the aid of special studies to detect viral antigens and/or nucleic acids in the blood or tissues. At times, it can also be difficult to distinguish acute rejection from acute hepatitis since both can contain a mononuclear portal infiltrate(1, 3). However, the primary focus of the necro-inflammatory activity in rejection is distinct from that associated with viral hepatitis. In acute hepatitis, the lobule is the focus of injury, which is manifest as spotty acidophilic necrosis of hepatocytes, lobular disarray. Although varying degrees of portal inflammation can be seen, significant damage to portal tract structures such as the bile ducts and veins is unusual. In contrast, the portal structures(portal vein and bile ducts) are the focus of immunologically mediated damage in acute rejection (1, 3).
It can be extremely difficult to distinguish between late onset acute or early chronic rejection from chronic hepatitis with low grade interface activity, particularly when no grand glass hepatocytes or sanded nuclei are seen. In such cases, a survey is taken of the percentage of damaged bile ducts. When there is damage of only an occasional bile duct and many bile ducts are un-inflamed, the cause of injury is more likely to be hepatitis. In contrast, rejection is more likely the cause of injury when there is damage of more than an occasional bile duct and a majority of the bile duct show degenerative changes such as eosinophilic transformation of the cytoplasm, uneven nuclear spacing and there is focal bile duct loss. In occasional cases it will be impossible to confidently distinguish between low grade acute rejection and chronic hepatitis on the basis of histopathology alone (45, 46). In such cases, our policy is to administer a bolus of corticosteroids and then follow the response to treatment over the next month. Typically, patients with rejection will show a more sustained response whereas those with viral hepatitis will transiently improve, but then relapse, with re-elevation of the liver injury tests after a period of a week or two. In some cases, rejection will primarily manifest as mononuclear inflammation in and around the terminal hepatic venules, which is often accompanied by perivenular hepatocyte dropout and sinusoidal red blood cell congestion. Even if the portal tracts show changes typical of hepatitis, in our experience, this so called "central venulitis" responds to increased immunosuppression (47).
Immunoperoxidase staining for HBV antigens is routinely carried out in almost all liver allograft biopsies from HBsAg+ recipients. In our experience, polyclonal antibody reagents are more sensitive but less specific than monoclonal anti-HBV reagents. Patients showing cytoplasmic HBcAg expression have experienced a more aggressive course in our experience (unpublished observation). However, detection of viral antigens does not mean that the liver inflammation and damage is due to the B virus(3). The final diagnosis should be based on the histopathological findings, including the pattern of injury.
REFERENCES
|
|
|
|